How COVID-19 damages lungs: The virus attacks mitochondria, continuing an ancient evolutionary battle

Viruses and bacteria have a very long history. Because viruses can’t reproduce without a host, they’ve been attacking bacteria for millions of years. Some of those bacteria eventually became mitochondria, synergistically adapting to life within eukaryotic cells (cells that have a nucleus containing chromosomes).
Ultimately, mitochondria became the powerhouses within all human cells.
Fast-forward to the rise of novel coronaviruses like SARS-CoV-2, and the global spread of COVID-19. Approximately five percent of people infected with SARS-CoV-2 suffer respiratory failure (low blood oxygen) requiring hospitalization. In Canada about 1.1% of infected patients (almost 46,000 people) have died.
This is the story of how a team, assembled during the pandemic, recognized the mechanism by which these viruses were causing lung injury and lowering oxygen levels in patients: It is a throwback to the primitive war between viruses and bacteria—more specifically, between this novel virus and the evolutionary offspring of bacteria, our mitochondria.
SARS-CoV-2 is the third novel coronavirus to cause human outbreaks in the 21st century, following SARS-CoV in 2003 and MERS-CoV in 2012. We need to better understand how coronaviruses cause lung injury to prepare for the next pandemic.
How COVID-19 affects lungs
People with severe COVID-19 pneumonia often arrive at the hospital with unusually low oxygen levels. They have two unusual features distinct from patients with other types of pneumonia:
- First, they suffer widespread injury to their lower airway (the alveoli, which is where oxygen is taken up).
- Second, they shunt blood to unventilated areas of the lung, which is called ventilation-perfusion mismatch. This means blood is going to parts of the lung where it won’t get sufficiently oxygenated.
Together, these abnormalities lower blood oxygen. However, the cause of these abnormalities was unknown. In 2020, our team of 20 researchers at three Canadian universities set about to unravel this mystery. We proposed that SARS-CoV-2 worsened COVID-19 pneumonia by targeting mitochondria in airway epithelial cells (the cells that line the airways) and pulmonary artery smooth muscle cells.
We already knew that mitochondria are not just the powerhouse of the cell, but also its main consumers and sensors of oxygen. Mitochondria control the process of programmed cell death (called apoptosis), and they regulate the distribution of blood flow in the lung by a mechanism called hypoxic pulmonary vasoconstriction.
This mechanism has an important function. It directs blood away from areas of pneumonia to better ventilated lobes of the lung, which optimizes oxygen-uptake. By damaging the mitochondria in the smooth muscle cells of the pulmonary artery, the virus allows blood flow to continue into areas of pneumonia, which also lowers oxygen levels.
It appeared plausible that SARS-CoV-2 was damaging mitochondria. The results of this damage—an increase in apoptosis in airway epithelial cells, and loss of hypoxic pulmonary vasoconstriction—were making lung injury and hypoxemia (low blood oxygen) worse.
Our discovery, published in Redox Biology, explains how SARS-CoV-2, the coronavirus that causes COVID-19 pneumonia, reduces blood oxygen levels.
We show that SARS-CoV-2 kills airway epithelial cells by damaging their mitochondria. This results in fluid accumulation in the lower airways, interfering with oxygen uptake. We also show that SARS-CoV-2 damages mitochondria in the pulmonary artery smooth muscle cells, which inhibits hypoxic pulmonary vasoconstriction and lowers oxygen levels.
Attacking mitochondria
Coronaviruses damage mitochondria in two ways: by regulating mitochondria-related gene expression, and by direct protein-protein interactions. When SARS-CoV-2 infects a cell, it hijacks the host’s protein synthesis machinery to make new virus copies. However, these viral proteins also target host proteins, causing them to malfunction. We soon learned that many of the host cellular proteins targeted by SARS-CoV-2 were in the mitochondria.
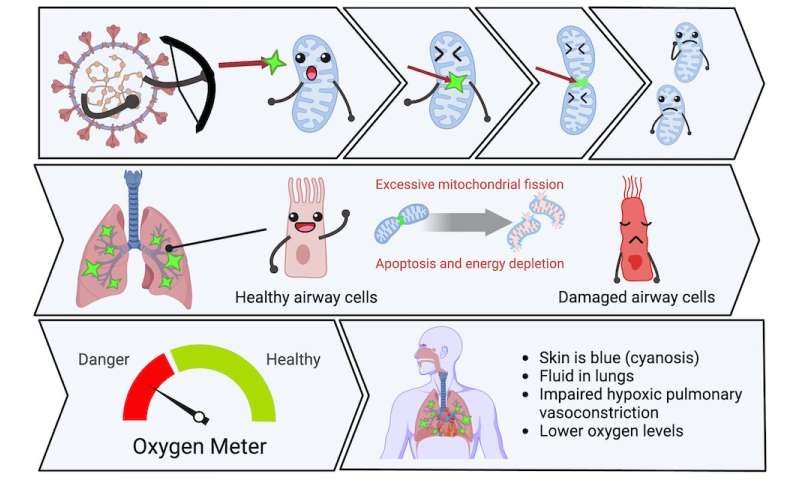
Viral proteins fragment the mitochondria, depriving cells of energy and interfering with their oxygen-sensing capability. The viral attack on mitochondria starts within hours of infection, turning on genes that break the mitochondria into pieces (called mitochondrial fission) and make their membranes leaky (an early step in apoptosis called mitochondrial depolarization).
In our experiments, we didn’t need to use a replicating virus to damage the mitochondria—simply introducing single SARS-CoV-2 proteins was enough to cause these adverse effects. This mitochondrial damage also occurred with other coronaviruses that we studied.
We are now developing drugs that may one day counteract COVID-19 by blocking mitochondrial fission and apoptosis, or by preserving hypoxic pulmonary vasoconstriction. Our drug discovery efforts have already enabled us to identify a promising mitochondrial fission inhibitor, called Drpitor1a.
Our team’s infectious diseases expert, Gerald Evans, notes that this discovery also has the potential to help us understand Long COVID. “The predominant features of that condition—fatigue and neurologic dysfunction—could be due to the lingering effects of mitochondrial damage caused by SARS-CoV-2 infection,” he explains.
The ongoing evolutionary battle
This research also has an interesting evolutionary angle. Considering that mitochondria were once bacteria, before being adopted by cells back in the primordial soup, our findings reveal an Alien versus Predator scenario in which viruses are attacking “bacteria.”
Bacteria are regularly attacked by viruses, called bacteriophages, that need a host to replicate in. The bacteria in turn fight back, using an ancient form of immune system called the CRISPR-cas system, that chops up the viruses’ genetic material. Humans have recently exploited this CRISPR-cas system for a Nobel Prize-winning gene editing discovery.
The ongoing competition between bacteria and viruses is a very old one; and recall that our mitochondria were once bacteria. So perhaps it’s not surprising at all that SARS-CoV-2 attacks our mitochondria as part of the COVID-19 syndrome.
Pandemic pivot
The original team members on this project are heart and lung researchers with expertise in mitochondrial biology. In early 2020 we pivoted to apply that in another field—virology—in an effort to make a small contribution to the COVID-19 puzzle.
The diverse team we put together also brought expertise in mitochondrial biology, cardiopulmonary physiology, SARS-CoV-2, transcriptomics, synthetic chemistry, molecular imaging and infectious diseases.
Our discovery owes a lot to our virology collaborators. Early in the pandemic, University of Toronto virologist Gary Levy offered us a mouse coronavirus (MHV-1) to work with, which we used to make a model of COVID-19 pneumonia. Che Colpitts, a virologist at Queen’s University, helped us study the mitochondrial injury caused by another human beta coronavirus, HCoV-OC43.
Finally, Arinjay Banerjee and his expert SARS-CoV-2 virology team at Vaccine and Infectious Disease Organization (VIDO) in Saskatoon performed key studies of human SARS-CoV-2 in airway epithelial cells. VIDO is one of the few Canadian centers equipped to handle the highly infectious SARS-CoV-2 virus.
Our team’s super-resolution microscopy expert, Jeff Mewburn, notes the specific challenges the team had to contend with.
“Having to follow numerous and extensive COVID-19 protocols, they were still able to exhibit incredible flexibility to retool and refocus our laboratory specifically on the study of coronavirus infection and its effects on cellular/mitochondrial functions, so very relevant to our global situation,” he said.
Source: Read Full Article