Blocking an ion channel improves muscle function and survival in mice with severe Duchenne muscular dystrophy
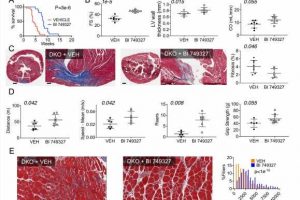
 Survival curves for DKO mice with BI 749327 treatment versus placebo (vehicle). (n=24 vehicle, n=15 BI 749327, log-rank test). Data in remaining panels are from ~6 wk old animals from each group. (B) Echocardiography: fractional shortening, cardiac output, and LV thickness. Unpaired t-test, n=7 vehicle (VEH) and BI 749327. (C) Left and Middle: Example 6-weeks Masson’s trichrome stains of whole heart cross sections with high-magnification of region focal fibrosis. Scale bar = 500 μm for whole heart section and 100 μm for 20x image. Right: Summary analysis, unpaired t-test; n=8 vehicle, n=6 BI 749327. (D) Integrated skeletal motor function assessed by open field test distance and speed (n=6 vehicle, n=7 BI 749327; unpaired T test), rearing behavior (n=5 vehicle, n=7 BI 749327, Mann Whitney), and grip strength (n=5 vehicle, n=10 BI 749327, Mann Whitney). (E) Example histology and summary distribution of cross-sectional area of muscle fibers in gastrocnemius (n=659 vehicle, n=563 BI 749327 fibers, n=3 animals/group, Mann Whitney). Credit: <i>Journal of Clinical Investigation—Insight</i> (2022). DOI: 10.1172/jci.insight.158906")
Researchers at Johns Hopkins Medicine report that an experimental drug first developed to treat kidney disease prolongs survival and improves muscle function in mice genetically engineered to develop a severe form of Duchenne muscular dystrophy (DMD).
According to the Muscular Dystrophy Association (MDA), DMD affects 1 in every 5,000 live male births and results in severe muscle wasting and weakness because of the lack of a protein called dystrophin, which is needed to strengthen muscle cells and protect them from mechanical injury. The dystrophin gene is in the X chromosome, so DMD mainly affects boys.
Girls only get the disease if both X chromosomes are affected. Muscle symptoms begin between the ages of 2 and 4, and by their early teens, most individuals with DMD are unable to walk. Dystrophin is also important to heart muscle, so heart failure often occurs in later teenage years into the early 20s. According to the MDA, people with DMD generally live until their late 20s or early 30s.
There is no cure, but physical therapy and corticosteroids reduce inflammation and help delay muscle decline, helping symptoms and improving quality of life. Newer gene-targeting treatments are being tested, but they remain limited to a small number of patients targeted.
The new study, published Sept. 13 in the JCI Insight, reports that a drug blocking the ion channel called TRPC6 in mice with severe DMD doubled their survival and improved their skeletal and cardiac muscle function. It also reduced bone deformities associated with weak muscles.
Ion channels are groups of proteins mostly found at the outer cell membrane that act like pores to allow elements like calcium or sodium to pass into or out of the cell in a controlled way. TRPC6 is one of these proteins that mostly allows calcium to pass into the cell.
It is activated by stress hormones and mechanical forces, and has been found to contribute to abnormal calcium entry into muscle cells lacking dystrophin. This can result in cell damage and ultimately cell death, according to David Kass, M.D., Abraham and Virginia Weiss Professor of Cardiology at the Johns Hopkins University School of Medicine and senior author of the study.
“In 2014, my laboratory first showed that calcium entering heart muscle cells from Duchenne mice rose more when they were stretched as compared to normal cells. This induced abnormal rhythms. We found that blocking TRPC6 prevented this excess calcium from entering the heart and reduced arrhythmia,” Kass says. “We thought maybe a long-term TRPC6 blocker would help with this syndrome. Unfortunately, the drug we had back then worked fine in cells but was metabolized too fast in animals, so it was not usable.”
For the new study, the researchers used a mouse model of DMD that mimics the severe disease found in humans. The mice had a median lifespan of two months (equal to late teens for a human). Starting three days after birth, researchers treated male and female DMD mice with a TRPC6 inhibitor drug (BI 749327, developed by Boehringer-Ingelheim Pharmaceuticals) or with a placebo. Mice receiving the placebo had a median survival rate of four weeks and 100% mortality by nine weeks. Mice getting the inhibitor had nearly double this lifespan.
Heart function improved by nearly 50%, and mice moved around 50% more and faster when compared with DMD mice that did not get the drug. These findings were supported by another experiment in which TRPC6 was genetically removed so the DMD mice never expressed the protein. They also had prolonged lifespan (nearly three times controls) and improved muscle function.
“We believe this may be one of the first studies to prolong survival in this very severe model of Duchenne with a compound that can be administered orally. While the molecule does not restore dystrophin, it blocks abnormal behavior of another protein that is caused by the lack of dystrophin,” says Kass. “This is not a cure for Duchenne, but if human trials replicate these findings, it could mean patients may survive into their late 40’s and 50’s with the possibility for an improved quality of life.”
Source: Read Full Article