Researchers expand and upgrade the 1000 Genomes Project resource using whole-genome sequencing
")
Seven years ago, the 1000 Genomes Project (1kGP) published an open-access resource based primarily on low-coverage whole-genome sequencing (WGS) data of 2,504 individuals from 26 populations representing five continental regions of the world, making it the first large-scale WGS effort to deliver a catalog of human genetic variation.
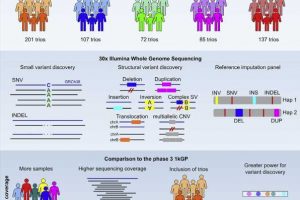
Now, researchers at the New York Genome Center (NYGC), in collaboration with groups at the Massachusetts General Hospital, Yale University, and Human Genome Structural Variation Consortium (HGSVC), have expanded the 1kGP resource to include nearly all parent-child trios in the collection, alongside the original samples, and sequenced them at high coverage using Illumina NovaSeq instruments. The study, published in Cell, presents comprehensive analyses of the high-coverage WGS data on the expanded 1kGP cohort which now consists of 3,202 samples, including 602 trios.
“The 1000 Genomes Project cohort is such a valuable resource, we felt it would be useful to the community to bring the sequencing up to date with the latest version of short-read technology while adding in the richness of the previously omitted family samples,” explained Michael Zody, Ph.D., Scientific Director of Computational Biology at the NYGC, and the study’s senior author.
Using state-of-the-art methods and algorithms, researchers at the NYGC sequenced DNA derived from lymphoblastoid cell lines (LCLs; i.e., immortalized human B cells from peripheral blood) from the expanded cohort to a targeted depth of 30X genome coverage. Next, the group performed single nucleotide variant (SNV) and short insertion and deletion (INDEL) calling, which consists of identification of variant sites from the sequence data relative to the human genome reference and genotyping of discovered variant sites across all samples in the cohort.
Additionally, a team from Dr. Michael Talkowski’s group at the Harvard Medical School, Broad Institute and Massachusetts General Hospital, in collaboration with Dr. Ira Hall’s group at Yale University and the Washington University School of Medicine, as well as the HGSVC, discovered and genotyped a comprehensive set of structural variants (SVs) across the 3,202 1kGP samples by integrating multiple analytic approaches.
Overall, the study shows significant improvements in both discovery power and precision of variant calls, especially among rare SNVs as well as INDELs and SVs spanning the frequency spectrum, which were previously inaccessible with low-coverage sequencing.
An important aspect of the original 1kGP resource is its use as a reference panel for variant imputation, i.e., statistical inference of unobserved genotypes in sparse, array-based samples based on groupings of variants that are typically inherited together in the population learned from the reference panel, which facilitated numerous genome-wide association studies (GWAS). Now, with the expansion of the original resource, the team upgraded the reference imputation panel to include more variants discovered through high-coverage WGS and trio families.
“The new imputation panel includes more sites, especially many more common INDELs and SVs, thus expanding the number of variants accessible for GWAS, which, given the large effect size of non-SNV variation, is likely to enable discovery of new genetic associations that help pinpoint the causative variant,” explains Marta Byrska-Bishop, Ph.D., Senior Bioinformatics Scientist at the NYGC, and the study’s co-first author.
All raw sequence data and variant call sets were immediately released to the public upon sequencing completion via several genomic data repositories, including the International Genome Sample Resource (IGSR) which is maintained by co-authors from the European Bioinformatics Institute at the European Molecular Biology Laboratory (EMBL-EBI).
“Our goal is to have this public resource serve as the benchmark for future population genetic studies and methods development,” adds Xuefang Zhao, Ph.D., Postdoctoral Fellow at the Center for Genomic Medicine Massachusetts General Hospital, and the study’s co-first author.
Source: Read Full Article