Narrowing down on small molecular inhibitors of SARS-CoV-2 key protease

A recent methodological study by researchers from the University of Luxembourg shows that the combination of virtual screening approaches, molecular dynamics simulations and machine learning can substantially facilitate small molecule screening investigations targeted at key SARS-CoV-2 viral proteins. Their findings are currently available on the bioRxiv* preprint server.
The coronavirus disease 2019 (COVID-19) pandemic, which is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is still a major challenge for healthcare systems and economies worldwide. And while vaccines are being rolled out all over the world, the available drug-based therapies are still lacking.
_-_1200_width.jpg)
Among such global biomedical efforts, one of the more salient and intensely studied pharmacological strategies is to significantly reduce viral load in infected individuals that have developed severe forms of the disease. From a biological standpoint, we now understand the virus well enough to identify the best potential targets for action.
One of them is the SARS-CoV-2 viral 3C-like protease (abbreviated as 3CLpro or Mpro), which represents an indispensable component for viral replication. This specific protease is utilized by the virus cleaving viral polyproteins into eleven non-structural proteins.
Ongoing computational screening endeavors that are ligand- and receptor-based would be made easier by an improved understanding of the hydrophobic, electrostatic and steric features that are specific for small molecule inhibitors which bind stably to 3CLpro, but are also valid for an extended collection of identified binders.
Consequently, Dr. Enrico Glaab, Dr. Ganesh Babu Manoharan and Dr. Daniel Abankwa from the University of Luxembourg aimed to identify small molecule inhibitors of 3CLpro with micromolar activity, as well as to develop a pharmacophore model that clearly delineates functional chemical groups associated with the molecular ligand recognition by the 3CLpro binding pocket.
Exploring potential protease inhibitors
In this study, top-ranked compounds and a handful of reference molecules from the literature that were previously proposed as potential 3CLpro inhibitors were experimentally appraised with the use of a ligand activity assay in order to identify a subset of inhibitors and their IC50 values (i.e., a concentration needed for 50% inhibition in vitro).
Based on the size of the compound libraries, the researchers tested distinct virtual screening methodologies – hence, also comparing and analyzing the utility of various strategies that can be used for inhibitor discovery.
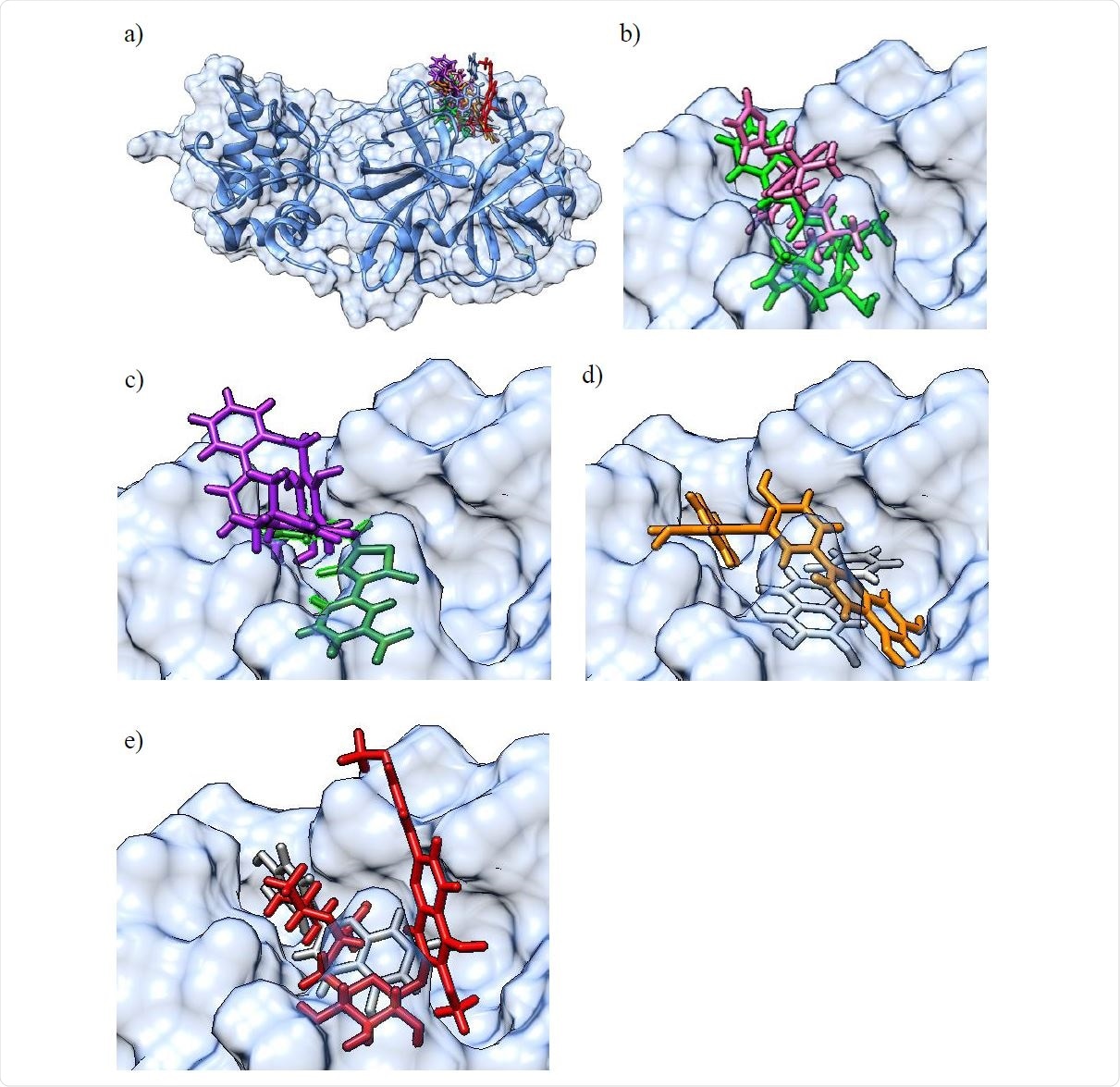
Finally, the validated inhibitors (including both natural compounds with existing information and previously uncharacterized synthetic ones) were used to develop a pharmacophore model, which is basically a computational model that elucidates chemical and steric features of compounds that can bind to the target protein.
Safe and bioavailable active compounds
This type of combined computational and experimental approach (as delineated in-depth in this paper) revealed new natural and synthetic compounds that can inhibit 3CLpro with micromolar activity (such as rottlerin and M-9420). Furthermore, the researchers have also provided a pharmacophore model that illustrates the most important structural and chemical properties of active compounds.
More specifically, detailed pharmacophore analyses indicate that many of the 3CLpro inhibitors share hydrogen bond and hydrophobic interactions with binding pocket residues, which include interactions with cysteine and histidine residues pivotal for catalysis.
Moreover, the analysis of absorption, distribution, metabolism, excretion, toxicity and physicochemical parameters of the confirmed hit compounds reveal that these compounds differ substantially in regards to known or computationally estimated properties that are deemed relevant. Still, a subset of the hits shows rather favorable bioavailability and safety traits.
Implications and further research
This study has shown that each of the used approaches uniquely identified at least one of the experimentally confirmed inhibitors, which implies that different screening strategies can provide complementary and independent information useful for increasing the number and diversity of identified active compounds.
Identifying structurally and chemically diverse active compounds with a similar binding mode is important for the creation of more comprehensive integrated pharmacophore models", emphasize the authors of this bioRxiv study.
Thus, now there is a need for extended screening and structural optimization analyses for 3CLpro inhibitors. This will help significantly not only in opening the door for additional activity appraisals in preclinical models, but also for subsequent lead compound development.
*Important Notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Glaab, E. et al. (2021). A pharmacophore model for SARS-CoV-2 3CLpro small molecule inhibitors and in vitro experimental validation of computationally screened inhibitors. bioRxiv. https://doi.org/10.1101/2021.03.02.433618, https://www.biorxiv.org/content/10.1101/2021.03.02.433618v1
Posted in: Medical Science News | Medical Research News | Disease/Infection News | Healthcare News
Tags: Assay, Compound, Coronavirus, Coronavirus Disease COVID-19, Cysteine, Healthcare, Histidine, in vitro, Ligand, Machine Learning, Metabolism, Molecule, Pandemic, Preclinical, Protein, Receptor, Research, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome, Virus

Written by
Dr. Tomislav Meštrović
Dr. Tomislav Meštrović is a medical doctor (MD) with a Ph.D. in biomedical and health sciences, specialist in the field of clinical microbiology, and an Assistant Professor at Croatia's youngest university – University North. In addition to his interest in clinical, research and lecturing activities, his immense passion for medical writing and scientific communication goes back to his student days. He enjoys contributing back to the community. In his spare time, Tomislav is a movie buff and an avid traveler.
Source: Read Full Article