Omicron analysis shows no clear evidence for increased virulence or lethality
In a recent study published on the medRxiv* preprint server, researchers conducted a genomic sequencing and epidemiological correlation analysis of the severe acute respiratory syndrome-associated coronavirus 2 (SARS-CoV-2) Omicron variant (B.1.1.529) to determine the functional impact and epidemiological advantage of this variant as compared to other strains of SARS-CoV-2.
Study: An in silico analysis of early SARS-CoV-2 variant B.1.1.529 (Omicron) genomic sequences and their epidemiological correlates. Image Credit: Adao / Shutterstock.com
Background
The SARS-CoV-2 Omicron variant was declared a variant of concern (VOC) due to a large number of genomic mutations present in its spike (S) protein and the host receptor-binding domains (RBD). After the emergence of the Omicron variant, a surge of coronavirus disease (COVID-19) cases was reported in many countries worldwide.
The emergence of a sublineage of the Omicron, BA.2, with slight variation in the mutations and limited information about the epidemiological characteristics of the SARS-CoV-2 Omicron variant, warrants the need for more studies.
About the study
In the present study, the authors performed an in silico and epidemiological correlation analysis of the SARS-CoV-2 Omicron variant. To this end, they determined the functional impact of Omicron variant mutations on host-virus interactions and the epidemiological advantage of Omicron over specific SARS-CoV-2 variants. The current study was focused on the current and most prevalent sublineage of Omicron BA.1.
The in silico analysis used 3,604 genomic sequences of the Omicron variant from 54 countries collected from the Global Initiative on Sharing All Influenza Data (GISAID) up to December 10, 2021. The data was used to determine the functional impact of the Omicron’s mutations on virulence, immune escape, and viral transmission. A three-dimensional (3D) structure of the Omicron spike protein with amino acid changes was generated using hCoV-19/Wuhan/WIV04/2019 as a reference strain for the mutational analysis.
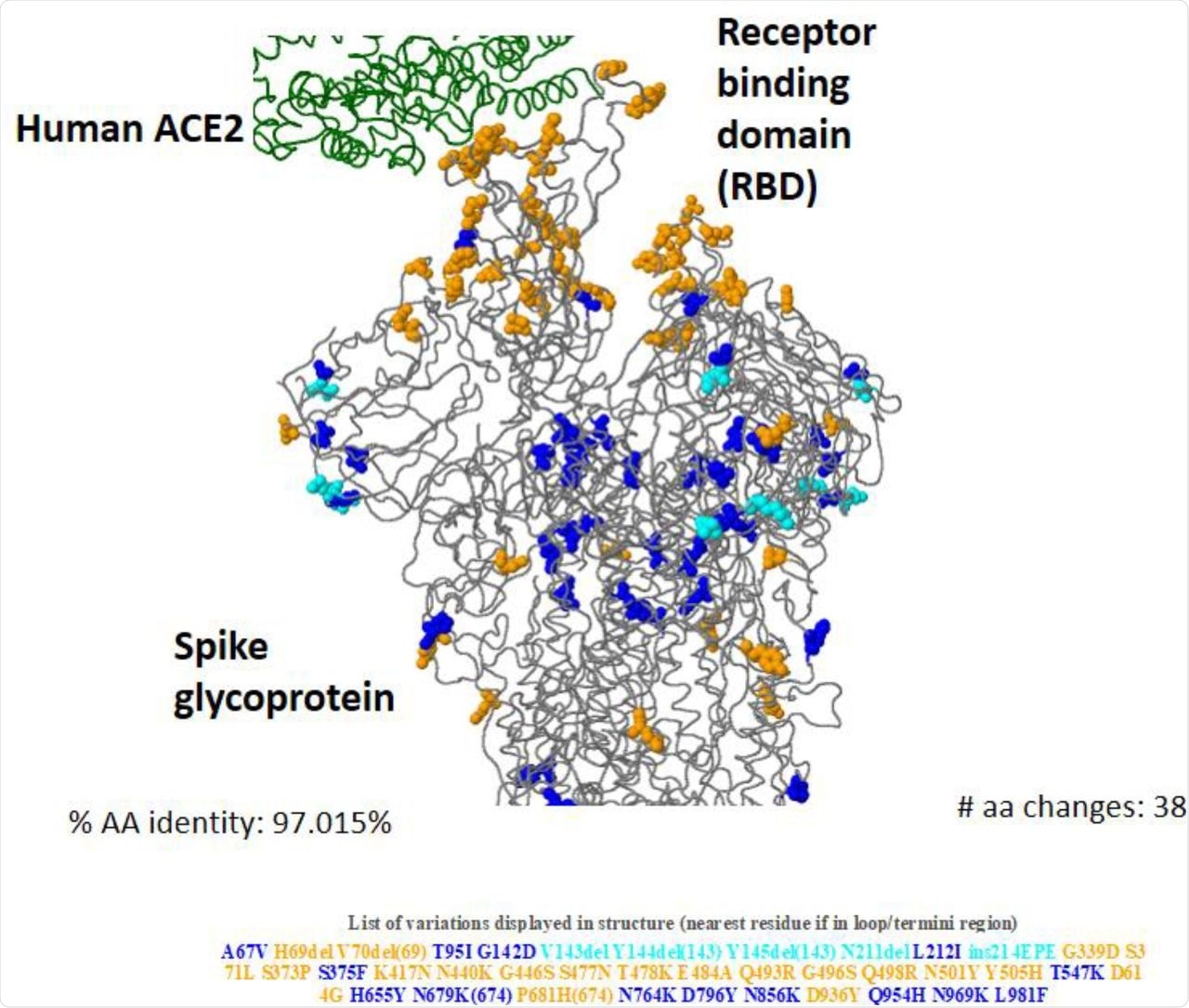
3D structure of Omicron (BA.1) spike glycoprotein in interaction of human ACE2 showing key amino acid substitutions.
RBD-associated mutations were determined using an open analysis pipeline that combined a yeast-display platform with deep mutational scanning. The pipeline determined the effect of amino acid mutations of the Omicron RBD in angiotensin-converting enzyme 2 (ACE2)-binding affinity and protein expression as compared to wild-type (WT) SARS-CoV-2 strain.
Subsequently, a comparative analysis between the relative proportion of genomic sequences of SARS-CoV-2 variants including Omicron and the recent COVID-19 cases and mortality data from SA was conducted from October 1, 2021, to December 10, 2021. This epidemiological correlation was performed to understand whether the Omicron variant has any epidemiological advantage in virulence or transmissibility as compared to existing SARS-CoV-2 variants. The global epidemiological data for the coronavirus disease 2019 (COVID-19) was collected from the Worldometer database.
Study findings
The results indicated that, as compared to other SARS-CoV-2 variants, Omicron had more genomic variation especially in the S protein region and receptor-binding motif (RBM). The Omicron variant had the closest complete sequence and RBM homology with the Alpha variant. Similar to the Alpha variant, Omicron also showed an S gene target failure (SGTF) during polymerase chain reaction (PCR) analysis.
About 81.5% of the total number of Omicron genomic sequences were reported from SA. The S protein sequence homology of the Omicron to the reference strain hCoV-19/Wuhan/WIV04/2019 ranged from 96.23 to 97.8% in the analyzed sequences. About 28 to 48 mutations were condensed in the S protein region of the Omicron variant and around 20 to 26 frequent non-S mutations were also noted.
In the mutational analysis, the RBD protein expression and ACE2-binding affinity were decreased due to a large number of mutations in the Omicron variant. The initial inverse correlation of the Delta and Omicron variant sequences reported from South Africa (SA) after the emergence of the Omicron variant later showed a decreasing trend. The Omicron mutations were categorized into host receptor binding (10), immune escape (20), virus replication (18), and host adaptability (3) groups.
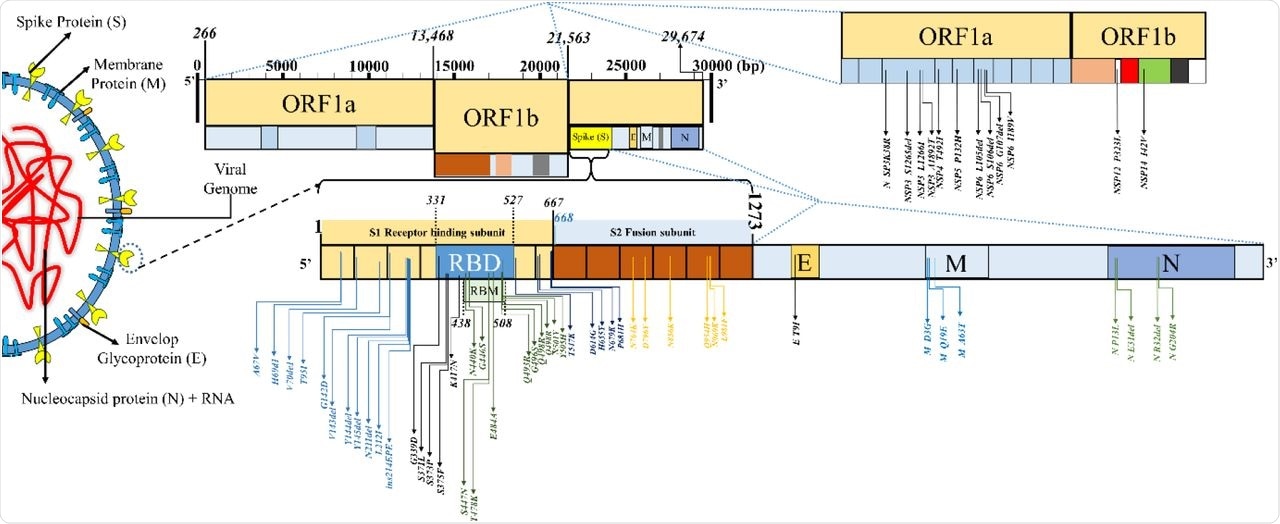
The mutational landscape in SARS-CoV-2 variant B.1.1.529 (Omicron, sublineage: BA.1).
There was about a 74% to 100% increase in new COVID-19 cases that were reported since the first Omicron case was identified. However, there was no associated increase in the incidence of new COVID-19-related deaths.
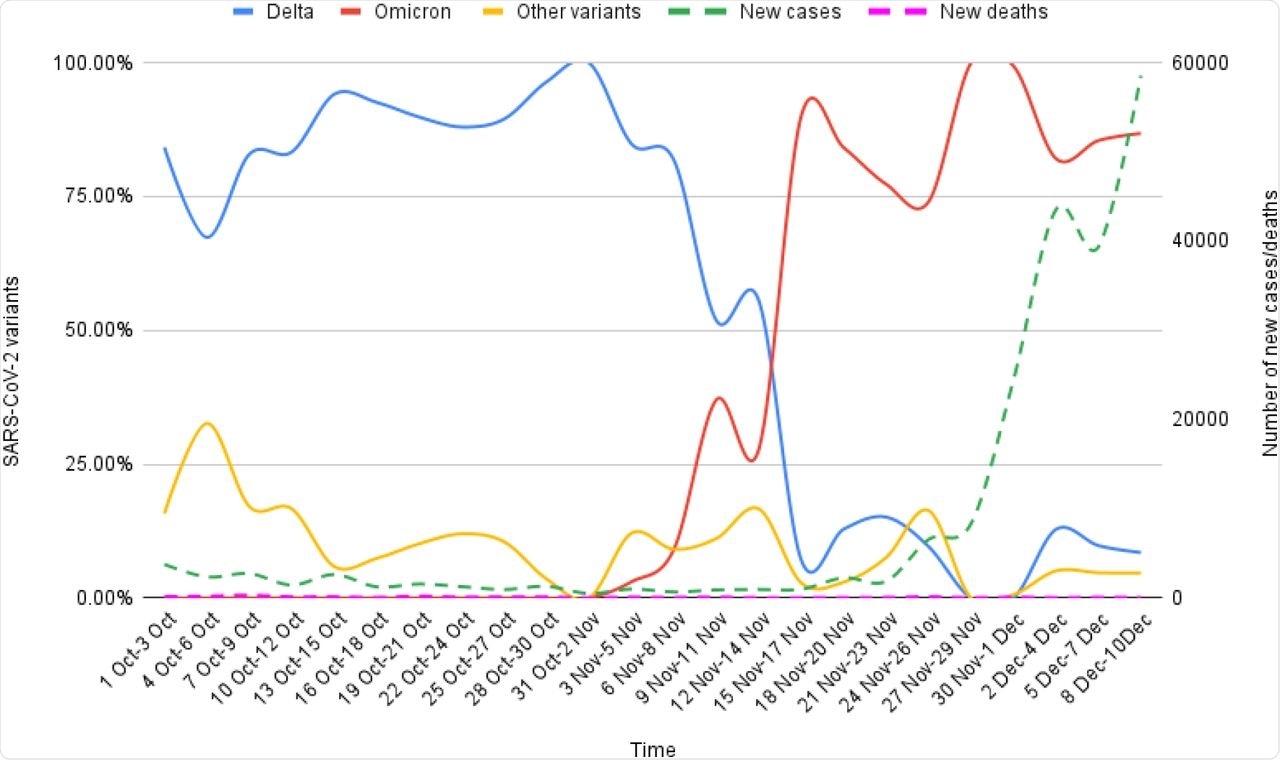
Epidemiological correlates of Omicron and Delta variants genomic sequences reported on GISAID from South Africa for the period of 1st Oct-10th Dec, 2021.
Conclusions
The findings concluded that the SARS-CoV-2 Omicron variant has increased immune evasion as compared to the other SARS-CoV-2 VOCs. There was no clear indication from the predictive mutational analysis that Omicron has increased virulence as compared to the other SARS-CoV-2 variants, including the Delta variant. Thus, the surge of Omicron cases in SA may be due to the increased immune escape capabilities of Omicron.
The results indicated that RBD mutations in the Omicron variant affected the ACE2 binding affinity, as well as protein expression. The host-virus interaction data derived from the study can be used to predict epidemiological potentials of the Omicron variant.
However, further studies about the dynamic interactions and allosteric influence of the RBD and other S protein region-associated mutations in Omicron are necessary to derive an exact impact on host-receptor binding and its clinical significance. Overall, the current study provides significant data for future research on the origin and epidemiological characteristics of the SARS-CoV-2 Omicron variant.
*Important notice
medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Kumar, A., Asghar, A., Singh, H. N., et al. (2021). An in silico analysis of early SARS-CoV-2 variant B.1.1.529 (Omicron) genomic sequences and their epidemiological correlates. medRxiv. doi:10.1101/2021.12.18.21267908. https://www.medrxiv.org/content/10.1101/2021.12.18.21267908v1.
Posted in: Genomics | Medical Science News | Medical Research News | Disease/Infection News
Tags: ACE2, Amino Acid, Angiotensin, Angiotensin-Converting Enzyme 2, binding affinity, Coronavirus, Coronavirus Disease COVID-19, Enzyme, Gene, Genomic, Genomic Sequencing, Glycoprotein, Influenza, Mortality, OCT, Polymerase, Polymerase Chain Reaction, Protein, Protein Expression, Receptor, Research, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Spike Protein, Syndrome, Virus, Yeast

Written by
Shanet Susan Alex
Shanet Susan Alex, a medical writer, based in Kerala, India, is a Doctor of Pharmacy graduate from Kerala University of Health Sciences. Her academic background is in clinical pharmacy and research, and she is passionate about medical writing. Shanet has published papers in the International Journal of Medical Science and Current Research (IJMSCR), the International Journal of Pharmacy (IJP), and the International Journal of Medical Science and Applied Research (IJMSAR). Apart from work, she enjoys listening to music and watching movies.
Source: Read Full Article