What are Prions?

Prions are proteins encoded by the PRNP gene that are composed of 253 amino acids. These proteins are found in two forms including the benign cellular form (PrPC) and the abnormal misfolded form (PrPSc).
Prions were first discovered in the 1980s during laboratory research on neurodegenerative diseases. Scientists noticed how prion proteins were highly expressed in nerve cells and that the abnormal PrPSc form acted as an infectious pathogen with the capacity to degenerate the central nervous system.
Neurodegenerative conditions caused by the presence of PrPSc are classified as prion diseases. Examples include sporadic Cruetzfeldt-Jacob disease (sCJD), Fatal familial insomnia (FFI), and iatrogenic CJD (iCJD). New diagnostic methods for prion diseases are currently being tested including identifying non-specific and specific biomarkers, as well as sequencing of the PRNP gene.
Furthermore, scientists are working to discover therapeutic strategies to treat prion diseases. The majority of which aim to reduce the level of abnormal PrPSc present to limit nerve damage.
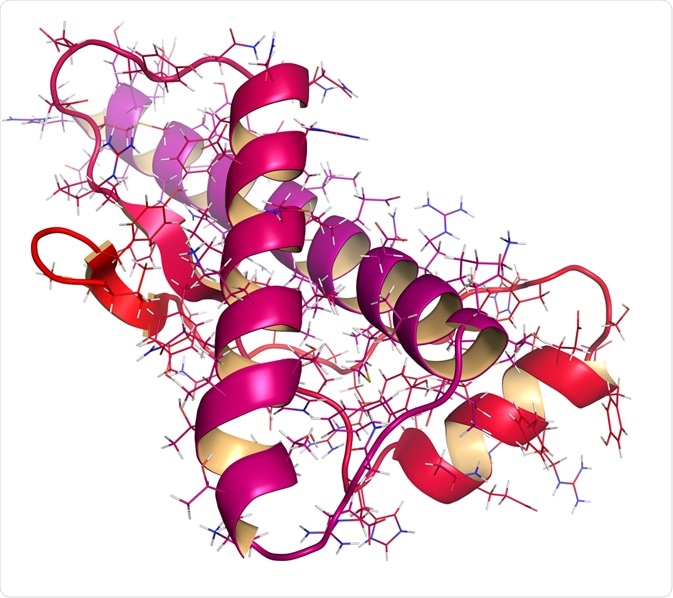
Image Credit: StudioMolekuul/Shutterstock.com
Prions and their structure
The normal cellular prion protein (PrPC) is converted into a misfolded, abnormal, disease-causing isoform (PrPSc) through a post-translational modification. Circular Dichroism (CD) and Nuclear Magnetic Resonance (NMR) spectroscopy studies confirm that the energy difference between the normal and abnormal states is very small, approximately 1-2 kcal mol-1. Thus, making such a transition from PrPC to PrPSc possible.
In terms of the primary amino acid sequence, both PrPC and PrPSc are identical. However, the post-translational alteration induces a change in the three-dimensional, folded conformation of PrPSc. PrPC is rich in an α-helical conformation with no β-sheet. In contrast, PrPSc is heavily composed of β-sheets with very few α-helical motifs present. The β-sheets in PrPSc can polymerize to form aggregated amyloid fibril structures that are insoluble and protease-resistant.
Last year, the first structure of human PrPSc fibrils were presented by cryo-EM. This study concluded that the aggregated prion fibrils are composed of a hydrophobic, protease-resistant core whilst the rest of the structure is entirely dominated by infinite B-sheets packed against each other. These fibrils are highly neurotoxic and therefore their formation leads to the development of prion disease.
Prion diseases
Prion diseases are fatal neurodegenerative conditions caused by the misfolding of PrPC into PrPSc which subsequently aggregates into fibrils within the brain. Prion diseases have a broad spectrum of clinical manifestations and therefore are subdivided into three main groups. This includes sporadic, genetic, and acquired forms.
Sporadic Cruetzfeldt-Jacob disease (sCJD) is a type of prion disease that has an annual worldwide incidence of 1-2 cases per million population. The presence of PrPSc distributed in the central nervous system is a hallmark feature found in sCJD patients. The most common age of onset is between 55 to 75 years and the disease displays equal prevalence in both males and females. Clinical symptoms of sCJD include cerebellar dysfunction, rapidly progressive dementia, behavioral changes, and tremors.
Fatal familial insomnia (FFI) is a genetic form of prion disease that is inherited in an autosomal dominant manner. FFI is caused by a mutation in the PRNP gene that encodes for prion protein. The most common FFI causing mutation is a point mutation at codon 178 of PRNP where aspartic acid is replaced with asparagine. Whilst this neurodegenerative prion disease is primarily characterized by difficulty with sleeping and brain damage, a wide range of other clinical symptoms can be seen such as muscle spasms and problems with memory and thinking.
Iatrogenic CJD (iCJD) is an acquired form of prion disease first described in 1974 in a patient who received a cadaveric corneal transplant from a donor with CJD. Since then, many cases of iatrogenic transmission of CJD have been identified. A significant proportion of iCJD cases were caused by human growth hormone (hGH) treatments. This was due to old pharmaceutical practices that involved processing 5,000 to 20,000 cadaveric pituitary glands in a single batch to extract hGH.
Many of these batches were contaminated with PrPSc from unknown CJD cases, thus causing 1 iCJD case per 100 patients receiving hGH treatment. Since 1987, the availability of recombinant hGH has led to a drastic reduction in iCJD cases recorded.
Diagnostic methods for prion diseases
Although histology of autopsy specimens remains the standard diagnostic tool for prion diseases, scientists are looking to utilize clinical laboratory testing for antemortem diagnosis. Thus, allowing patients to prepare to live with their disease and allowing the possibility of treatment to be provided to slow the progression of neurodegeneration with future medicine.
Cerebrospinal fluid (CSF) analysis can be used as a diagnostic tool to test for non-specific biomarkers of prion disease. Although they do not detect PrPSc directly, certain non-specific biomarkers are often elevated in prion diseases and can provide evidence for rapid neurodegeneration.
For example, the protein 14-3-3 in CSF can be measured to indicate neuronal injury. This was the first well-described prion disease marker to be identified. Another biomarker is Tau protein which is involved in microtubule stabilization within neurons and found in high concentrations in the CSF of patients with prion disease. Other non-specific biomarkers include neuron-specific enolase (NPE), S100B, and alpha-synuclein.
Measurements of these analytes in the CSF of patients can be achieved through routine immunoassays. Typically, enzyme-linked immunosorbent assays (ELISAs) are used to obtain a quantitative measurement of the non-specific biomarkers; whilst western blotting is used for qualitative analysis. Although measuring these markers can be used to diagnose prion diseases, extracting bloody CSF samples from patients can lead to false-positive or inaccurate results.
Other diagnostic methods for prion diseases that directly detect the presence of PrPSc have also been developed. The most conventional method involves analysis of brain tissue for levels of PrPSc. This requires protease K treatment and subsequent western blotting. Normal cellular PrPC would be degraded by protease K treatment and therefore would be undetectable by western blotting.
On the other hand, since PrPSc is protease-resistant it remains intact throughout the treatment and is detectable by western blotting. Although this test is specific to prion disease, it can have low sensitivity as PrPSc is typically distributed unevenly throughout the brain.
For genetic sub-types of prion diseases, nucleic acid sequencing of the PRNP gene can be performed to identify particular mutations strongly associated with the disease. This method of diagnosis is becoming increasingly popular for a wide variety of diseases as greater knowledge of genetics is attained. Sequencing the PRNP gene can be achieved using the peripheral blood of the patient. With ongoing sequencing studies, a greater number of mutations with a predisposition for prion diseases will be discovered to utilize for diagnosis.
Therapeutic strategies to treat prion diseases
With increasing knowledge of prion diseases over the years, scientists have begun to dedicate research into discovering novel therapeutic approaches to treat patients. Most treatments being tested on humans involve using drugs to reduce the conversion of PrPC to PrPSc to limit the presence of aggregated fibrils. Moreover, other studies are focusing on designing and generating antibodies that can recognize and bind onto the fibrillar PrPSc, thus creating a strong immune response against the neurotoxic disease-causing fibrils.
Unfortunately, many clinical trials so far have been unsuccessful. This is mainly due to the challenges faced in conducting clinical trials for prion disease. Many cases of prion disease are diagnosed when the disease has significantly progressed whilst the rapid progression means that patients may no longer be able to participate in the trial as their condition worsens. Moreover, the geographic dispersal of patients with prion disease makes it difficult to arrange single-site clinical trials with adequate sample size.
Despite the unsuccessful trials, many lessons have been learned that can inform future clinical trials in the search for effective treatment modalities for patients with prion disease.
References:
- Appleby, B., Conner, A., Wang, H. (2019). Therapeutic Strategies for prion disease: a practical perspective. Current Opinion in Pharmacology. 44: 15-19.
- Baldwin, K., Correll, C. (2019). Prion Disease. Semin Neurol. 39: 428-439.
- Conner, A., Wang, H., Appleby, B., Rhoads, D. (2019). Clinical Laboratory Tests Used to Aid in Diagnosis of Human Prion Disease. Journal of Clinical Microbiology. 57: 1-12.
- Glynn, C., Sawaya, M., Ge, P., Gallagher-Jones, M., Short, C., Bowman, R., Apostol, M., Zhou, Z., Eisenberg, D., Rodriguez, J. (2020). Cryo-EM structure of a human prion fibril with a hydrophobic, protease-resistant core. Nature Structural & Molecular Biology. 27: 417-423.
- Imran, M., Mahmood, S. (2011). An overview of human prion diseases. Virology Journal. 8: 1-9.
- Prusiner, S. (1998). Prions. Proc. Natl. Acad. Sci. 95: 13363-13383.
- Prusiner, S. (2001). Neurodegenerative diseases and prions. The New England Journal of Medicine. 344: 1516-1526.
Last Updated: Nov 23, 2021

Written by
Naveen Dha
Naveen graduated from King’s College London where she attained a Bachelor of Science in Biochemistry. Within this course, she chose to study topics pertaining to the biology of cancer, molecular immunology, molecular biology, and protein structure. Throughout her degree, she partook in writing various practical proposals, reports, and literature reviews whilst also gaining multifaceted laboratory and research experience. It was through these projects that Naveen discovered her interest for scientific writing as it allowed her to remain intellectually curious, creative, and detail-orientated.
Source: Read Full Article