The importance of gene position for muscle development and integrity

Mutations in the nuclear structural protein lamin A produce rare, tissue-specific diseases called laminopathies. To study these diseases, researchers from the Gasser group introduced a mutation inducing a human laminopathy in C. elegans and monitored its effect on chromatin. Not only did they understand the molecular basis of the disease, they found a way to counteract the dominant defects of the mutation, suggesting a novel therapeutic pathway.
Laminopathies are a set of human diseases that lead to differentiated tissue dysfunction, with muscle being most commonly affected. While rare, these conditions have phenotypes that could provide insight into more common disorders, including muscular dystrophy, insulin resistance and aging. In laminopathies, a mutation is carried by the protein lamin A, or sometimes a protein that interacts with lamin A. Lamins are part of the nuclear lamina, a semi-rigid meshwork of proteins that ensures the shape of the cell nucleus and influences the spatial distribution of genomic domains. Its constituents are some of the most conserved proteins in the animal kingdom.
In differentiated animal cells, it is essential that the correct genes for a given tissue are expressed and the incorrect ones kept silent, to ensure that our organs and tissues maintain their identity and function. Improper gene expression is often correlated with human degenerative disease or cancer. The nuclear lamina plays a role in maintaining tissue integrity by sequestering heterochromatin (the compacted, transcriptionally silent fraction of the genome) at the edge of the nucleus.
https://youtube.com/watch?v=wuWHkkNqHv0%3Fcolor%3Dwhite
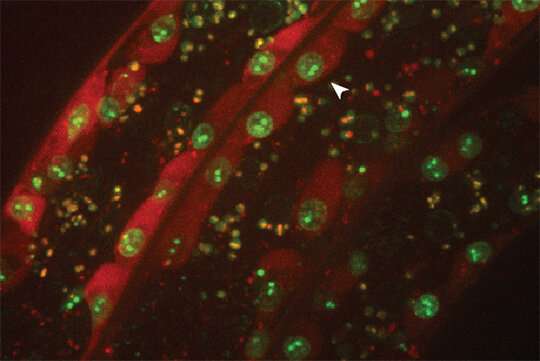
The Gasser group has studied this anchoring process for over a dozen years, making use of the model organism C.elegans. This small worm is ideal for the study of genome organization because it is fully transparent, allowing scientists to visualize proteins, genes and chromatin position by fluorescence microscopy. Studies in C.elegans revealed that heterochromatin is tethered at the nuclear lamina thanks to a specific chemical modification borne on the nucleosomes. This modification, histone H3K9 methylation, is recognized by CEC-4, a factor that is bound at the nuclear rim.
In a recent study, published in Genes & Development, two postdoctoral fellows in the Gasser lab, Jennifer Harr and Anna Mattout, focused on a human disease mutation in lamin A, which they introduced into C. elegans. This mutation causes a degenerative muscular dystrophy called Emery-Dreifuss Muscular Dystrophy, whereby the integrity of striated muscle is lost over time. The worms carrying the human mutation similarly lost the integrity of their body muscle, no longer swam normally and lost proper muscle ultrastructure. These muscular dystrophy-like phenotypes were correlated with an inappropriate distribution of muscle specific genes in the nucleus. Specifically, instead of shifting to an active, internal compartment of the nucleus, the muscle genes remained anchored at the nuclear lamina, as they are in other tissues where they are not expressed.
The researchers then set out to prove that the improper positioning of the muscle genes was the source of the disease symptoms. They disrupted the gene encoding CEC-4, the protein that anchors heterochromatin. The loss of this protein was sufficient to suppress all the phenotypes correlated with the mutant lamin: Normal muscle function and ultrastructure were restored; the worms swam like healthy worms. Looking at the genome organization in detail, Harr and collaborators found that the active muscle genes were released from the nuclear periphery. For many genes, but not all, this meant that the muscle appropriate gene expression pattern was restored. These results argue that the lamin-induced sequestration or retention of tissue specific genes in a heterochromatic compartment is the likely driver of the laminopathic disease.
Source: Read Full Article